

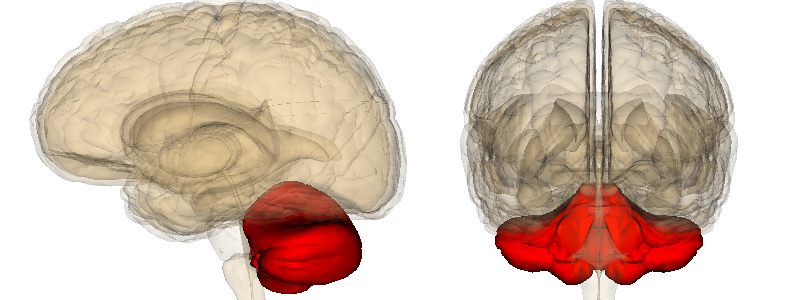




Giovanni Stevanin, researcher at the Brain and Spine Institute (Institut du Cerveau – ICM), and his collaborators identified a recurring mutation in a new gene responsible for cerebellar axatia that en-codes a calcium channel expressed in certain neurons of the cerebellum.
Numerous treatments that regulate the activity of calcium channels are widely used, and this discovery could lead to therapeutic advances for these rare diseases. This study was published in The American Journal of Human Genetics.
Cerebellar ataxias are degenerative disorders that affect the cerebellum, an important structure of the central nervous system that regulates balance and coordination. Patients suffering from this disorder present debilitating symptoms, handicapped walking, and im-paired movements. Certain forms of cerebellar ataxia have a genetic origin. However, even though more than 50 related genes have been discovered, the underlying cause for the dis-order is still unknown for more than 40% of patients.
Thanks to a combination of new high-throughput sequencing techniques and large-scale genome mapping, Giovanni Stevanin and Marie Coutelier at the Brain and Spine Institute (Institut du Cerveau – ICM), Philippe Lory at the Institute for Functional Genomics (IGF) in Montpellier, and their collaborators identified a recurring mutation in a new gene that affected three independ-ent families. This gene, CACNA1G, is responsible for autosomal dominant forms of cerebel-lar ataxia. The gene encodes for a channel, Cav3.1, that allows calcium ions to enter Purkinje cells in the cerebellum. The mutation affects a critical part of the protein that is responsible for detecting changes in the membrane potential of the cell that determines when the channel will open and close. The mutation described makes the channel less sen-sitive and reduces its overall activity.
These results bring attention back to the importance of ion channels in the cellular pro-cesses that lead to cerebellar ataxia. Interestingly, mutations that increase activity of the channel have already been described as being risk factors for epilepsy, a relationship that will be important to explore further in order to better understand these two families of pathologies. The frequent similarities between symptoms of epilepsy and ataxias linked to ion channel mutations illustrates the simultaneous involvement of ion channels in several complex neuronal processes.
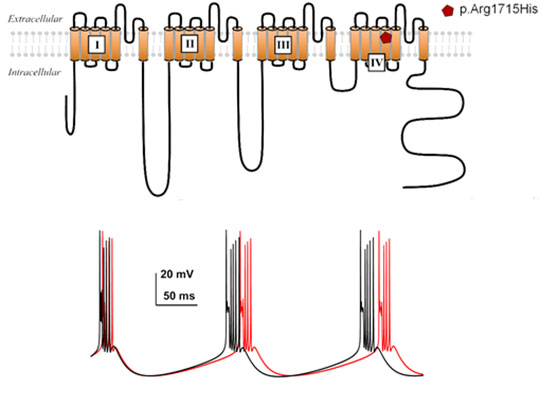
Figure. Top: schematic representation of the channel imbedded in the plasma membrane, showing the position of the recurring mutant identified in families with ataxia. Bottom: Stimulation of bursts of action potential burst discharges of deep neurons in the cerebel-lum, developed with the help of a NEURON simulation, shows the difference in discharge activity when the mutation is present (red) compared to the wild type channel (black).
Référence : A recurrent mutation in CACNA1G alters Cav3.1 T-type calcium channel con-duction and causes autosomal dominant cerebellar ataxia. Marie Coutelier et al., American Journal Human Genetics. 2015.