





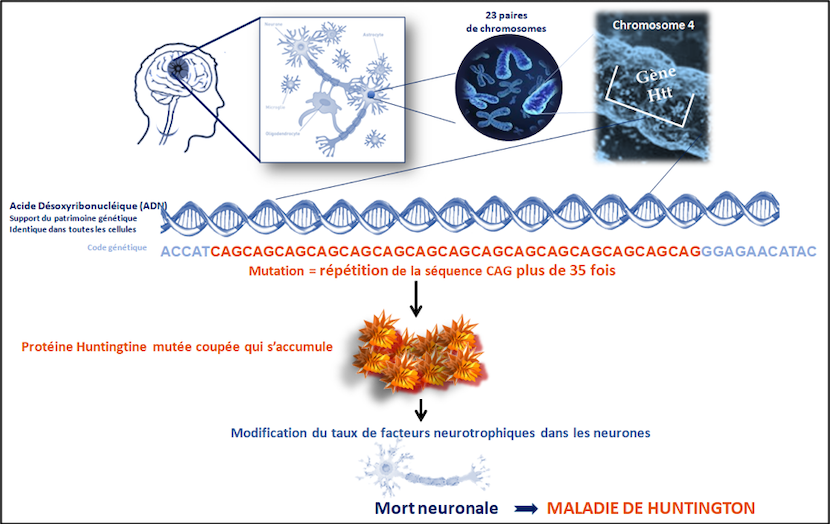
L’expansion est constituée d’une répétition anormale d’un triplet de nucléotides CAG dans l’ADN au niveau du chromosome 4. Une copie de la protéine devient anormale probablement toxique pour les cellules lorsque le nombre de répétitions CAG est supérieur à 35. La protéine huntingtine, dont le rôle essentiel est de permettre la survie des neurones ne suffit pas pour empêcher la maladie de se manifester. Les parties touché par l’atrophie au cours de la maladie sont le cortex et le striatum, zones cérébrales impliquées dans les fonctions motrices, cognitives et comportementales.
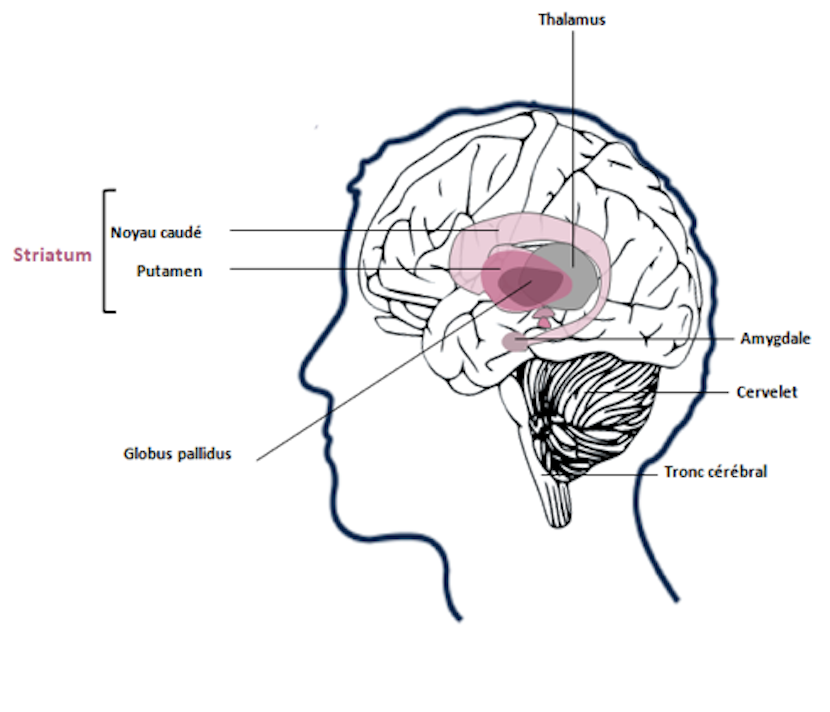
La maladie de Huntington touche le cortex et le striatum, impliqués dans les fonctions motrices, cognitives et comportementales.
A l’Institut du Cerveau
L’étude de la phase asymptomatique de la maladie de Huntington est un axe majeur de l’équipe « Neurogénétique fondamentale et translationnelle» codirigée par le Pr Alexandra Durr. En effet, les patients atteints de la maladie de Huntington sont nés avec l’expansion, pourtant ils ne développent les symptômes que des dizaines d’années plus tard. Que se passe-t-il entre temps ?
Une collaboration active entre la clinicienne-chercheuse et le Dr Sandrine Humbert au GIN à Grenoble qui a montré que des anomalies cérébrales étaient déjà présents à l’état fœtal. En revanche, la maladie ne se manifeste pas dès la naissance.
Il y a donc des signes d’atteinte cellulaire chez le fœtus, mais aucune atteinte clinique chez l’enfant et l’adolescent et les signes cliniques apparaissent à l’âge adulte. Il semble donc exister une compensation transitoire des déficits. L’objectif de l’équipe d’Alexandra Durr est de comprendre les mécanismes mis en jeu pendant la période non symptomatique de la maladie dans l’espoir de pouvoir activer les processus de compensation pour retarder l’apparition de la maladie.
Pour en savoir plus : https://institutducerveau-icm.org/fr/actualite/maladie-de-huntington-des-anomalies-cerebrales-stade-embryonnaire/