





DESCRIPTION DE LA MALADIE DE CHARCOT
La maladie de Charcot (SLA) est la maladie du motoneurone la plus fréquente chez l’adulte.
Il existe deux types de motoneurones :
- Les neurones moteurs centraux, situés dans une région particulière de notre cerveau, le cortex moteur, transmettent les ordres de contraction jusqu’à la moelle épinière.
- Les neurones moteurs périphériques, motoneurones situés dans la moelle épinière, transmettent l’information motrice jusqu’aux muscles.
La dégénérescence de ces 2 types de motoneurones au cours de la SLA entraine la perte de la transmission d’informations entre le cerveau et les muscles volontaires qui ne sont donc plus sollicités, ne se contractent pas et s’atrophient.
L’atteinte du motoneurone peut parfois être associée à une perte de neurones dans les régions frontales et temporales du cerveau caractéristiques, pouvant conduire à troubles cognitifs et comportementaux d’intensité variable. Dans sa forme la plus sévère, cela conduit à une démence fronto-temporale (DFT) : 15% des patients atteints de d’une maladie de Charcot sont également atteints de DFT. La démence dans la DFT ne se caractérise pas, comme dans la maladie d’Alzheimer, par des troubles de la mémoire, mais par des modifications comportementales qui sont au premier plan. Cela se traduit par des changements dans les comportements sociaux et de la personnalité des patients.
LES CAUSES DE LA MALADIE DE CHARCOT, EST-ELLE HEREDITAIRE ?
Pour 1 patient sur 10, l’origine de la maladie est due à des mutations génétiques héréditaires, on parle de cas familiaux.
Retrouvez plus d’informations et les recherches de l’Institut du Cerveau – ICM sur la page dédiée aux causes de la maladie de Charcot
LES MECANISMES BIOLOGIQUES DE LA MALADIE DE CHARCOT :
La maladie de Charcot ou sclérose latérale amyotrophique (SLA) est une maladie touchant les neurones moteurs, les cellules nerveuses qui commandent les mouvements volontaires. Ces motoneurones dégénèrent progressivement sans que l’on ne connaisse encore aujourd’hui les mécanismes précis conduisant à cette mort neuronale. Cette maladie neurodégénérative évolue en quelques années jusqu’à une paralysie complète.
Retrouvez plus d’informations et les recherches de l’Institut du Cerveau – ICM sur la page dédiée aux mécanismes biologiques de la maladie de Charcot
LES SYMPTOMES, LA PROGRESSION ET L’ESPERANCE DE VIE DE LA MALADIE DE CHARCOT :
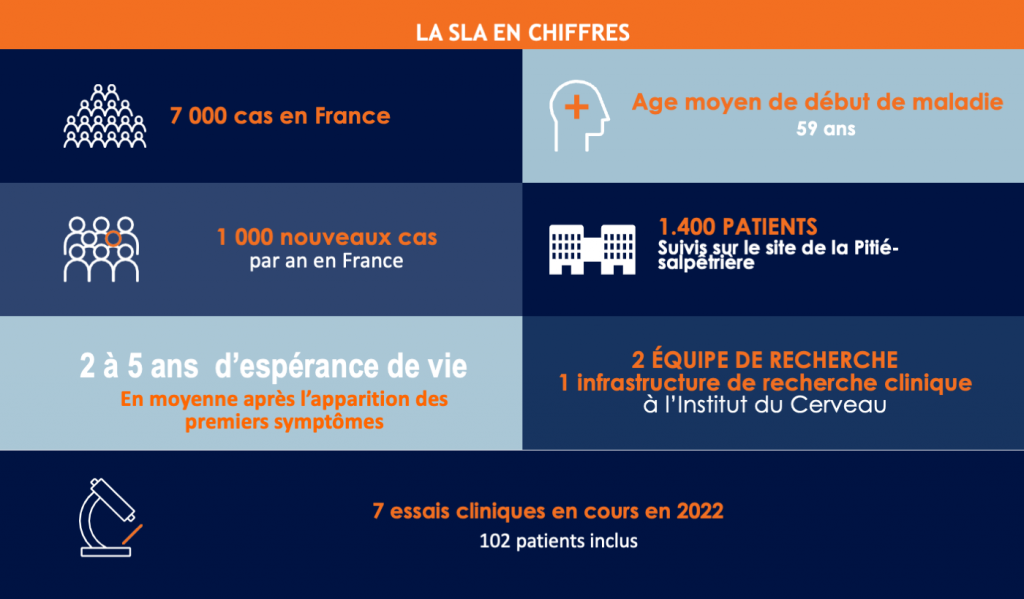
Les symptômes de la sclérose latérale amyotrophique (SLA) ou encore maladie de Charcot se caractérisent par une paralysie complète des muscles des bras, des jambes et de la région de la bouche et de la langue, ainsi que des muscles respiratoires. Cela entraine une incapacité à se servir de ses bras, marcher, manger, parler et des difficultés respiratoires qui s’installent progressivement. Cette maladie se déclare à l’âge adulte (moyenne à 59 ans en France) et va évoluer, en moyenne en 3 à 5 ans, vers la paralysie complète des muscles et conduire finalement au décès du patient, généralement par insuffisance respiratoire. En fonction de la localisation des neurones moteurs touchés : les neurones corticaux, du tronc cérébral et/ou de la moelle épinière, les symptômes présentés peuvent varier entre les différents patients, notamment en début de maladie.
Retrouvez plus d’informations et les recherches de l’Institut du Cerveau – ICM sur la page dédiée aux symptômes de la maladie de Charcot
QUELS SONT LES TRAITEMENTS ? COMMENT DIAGNOSTIQUER LA MALADIE DE CHARCOT ?
Le diagnostic de la maladie est suspecté par un neurologue qui retrouve à l’examen neurologique et à l’interrogatoire des signes et des symptômes témoignant d’une atteinte progressive du premier et deuxième motoneurone. Cela se caractérise dans tous les cas par une perte progressive de la force musculaire, parfois associé à une fonte musculaire, ou une raideur des membres. Pour poser ce diagnostic, le neurologue s’appuie sur les résultats d’un examen qui permet d’enregistrer les signaux électriques entre les neurones et les muscles : un électromyogramme. Celui-ci atteste de la dénervation motrice des muscles, conséquence de la perte des neurones moteurs. Afin de confirmer ce diagnostic, il est indispensable que le neurologue puisse écarter, par le biais d’autres examens complémentaires, toutes les autres causes possibles de souffrance de neurones moteurs telles que les compressions mécaniques, les atteintes inflammatoires des nerfs moteurs ou tout autre processus pouvant endommager les nerfs.
Dans le cas des formes familiales (10%) le diagnostic est établi de façon certaine lorsque le patient porte une des mutations connues, responsables de la maladie. Il y aujourd’hui plus de 30 gènes connus, et encore au moins 1/3 des cas familiaux n’ont pas de gènes identifiés (la proportion varie selon les régions du monde).
Il n’existe à ce jour aucun traitement curatif de la maladie de Charcot, cependant, l’association d’un traitement neuroprotecteur et d’une prise en charge multidisciplinaire, permettant une prise en charge des différents aspects de la maladie, permet de ralentir la progression des symptômes.
Retrouvez plus d’informations et les recherches de l’Institut du Cerveau – ICM sur la page dédiée au diagnostic de la maladie de Charcot
Dernière mise à jour juin 2022.