

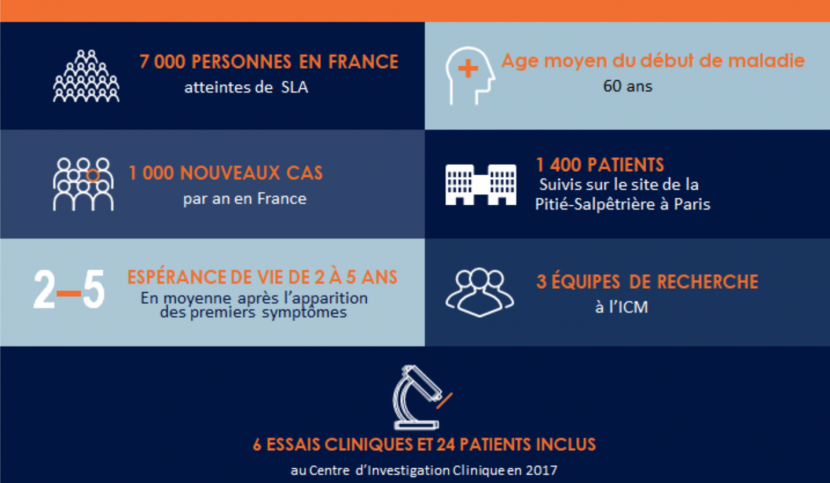




A l’occasion de la journée mondiale de la sclérose latérale amyotrophique (SLA) ou maladie de Charcot, l’Institut du Cerveau – ICM vous informe pour comprendre les mécanismes et les symptômes de cette maladie et vous parle des recherches de l’Institut dans ce domaine.
La Sclérose Latérale Amyotrophique (SLA) ou « Maladie de Charcot » est une pathologie neuromusculaire progressive et fatale caractérisée par une perte des motoneurones, neurones qui commandent entre autres la marche, la parole, la déglutition et la respiration. Il existe deux types de motoneurones :
- Les neurones moteurs centraux, situés dans une région particulière de notre cerveau, le cortex moteur, transmettent les ordres de contraction jusqu’à la moelle épinière.
- Les neurones moteurs périphériques, motoneurones situés dans la moelle épinière, transmettent l’information motrice jusqu’aux muscles.
L’atteinte neuronale entraine la disparition de la transmission d’informations entre le cerveau et la moelle épinière ou la moelle épinière et les muscles qui ne sont donc plus sollicités et s’atrophient.
Les causes :
10% des cas de SLA sont dits « familiaux » d’origine génétique. A ce jour 4 gènes majeurs mutés ont été identifiés comme responsables de la maladie. Le 1e gène considéré comme responsable de certaines SLA fut le gène de la superoxide dismutase 1 (SOD1) identifié en 1993 ; à ce jour plus de 150 mutations ont été décrites sur ce gène. Depuis, des mutations ont été identifiées sur les gènes TARDBP, FUS et C9ORF72 et les mutations dans les gènes majeurs concernent désormais près de deux tiers des SLA familiales. Il existe également des mutations identifiées dans une vingtaine d’autres gènes plus rares.
Dans les formes sporadiques de SLA, il existe des facteurs génétiques de prédisposition, c’est-à-dire des facteurs génétiques qui augmentent le risque de développer la maladie. On considère que la maladie est multifactorielle et que certains facteurs environnementaux ou liés au mode de vie des patients pourraient également contribuer au déclenchement de la maladie chez des individus prédisposés.
A L’Institut du Cerveau – ICM
Stéphanie MILLECAMPS dans l’équipe de Séverine BOILLEE cherche à identifier les mutations génétiques à l’origine de la maladie. Grâce à la participation des différents centres de référence de la SLA qui existent en France, les chercheurs ont pu rechercher les mutations génétiques de 400 familles touchées par la SLA et identifier la cause génétique de la maladie dans 70 % des cas. Ces études permettent de proposer un diagnostic moléculaire aux familles qui le souhaitent, mais aussi d’inclure des patients porteurs d’une mutation particulière dans les essais cliniques adaptés. Il reste encore 30% des cas familiaux à associer à des mutations causales et des approches d’analyses génétiques à grande échelle sont en cours pour y parvenir.
Un travail collaboratif européen a permis l’identification d’un nouveau gène impliqué dans la SLA. Nommé TBK1, il est impliqué dans l’élimination des déchets à l’intérieur des cellules et dans la régulation de l’inflammation, ce qui démontre l’importance des cellules immunitaires dans le déclenchement et probablement l’évolution de la maladie. Christian LOBSIGER dans l’équipe de Séverine BOILLEE (en collaboration avec une équipe allemande), modélise la SLA liée à des mutations dans le gène TBK1, pour étudier ces mécanismes et mieux comprendre la maladie.
Isabelle LE BER dans l’équipe d’Alexandra DURR et de Giovanni STEVANIN a participé à une étude internationale ayant identifié en 2018, un nouveau gène causal dans la SLA, la protéine KIF5A qui était d’ores et déjà en cause dans une autre pathologie du motoneurone central : la paraplégie spastique.
Les mécanismes :
Même dans les cas familiaux où les mutations responsables de la maladie ont été identifiées, le mécanisme conduisant à la dégénérescence des motoneurones n’est pas encore élucidé.
On observe une accumulation anormale de protéines incorrectement éliminées directement dans les motoneurones de la moelle épinière des patients. Ces analyses sont réalisées par le Pr Danielle SEILHEAN (service de Neuropathologie de la Pitié-Salpêtrière) et membre de l’équipe de Séverine BOILLEE. Le don de tissus post-mortem pour la recherche est une aide particulièrement précieuse pour les chercheurs de l’Institut du Cerveau – ICM.
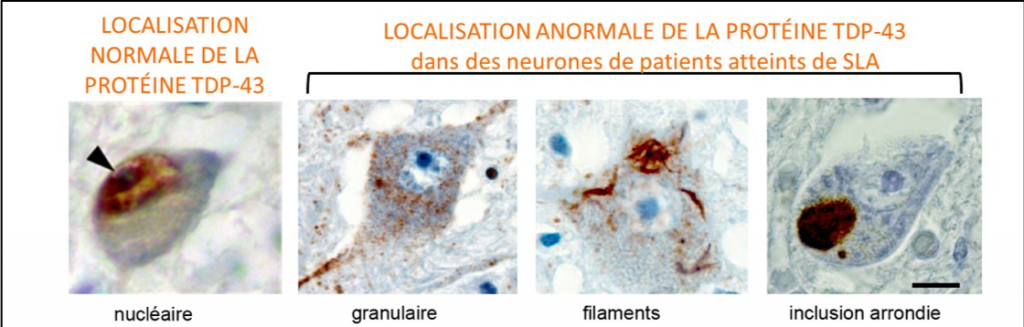
A l’Institut du Cerveau – ICM
L’équipe de Severine BOILLEE, étudie l’influence des mutations sur les cellules neurales ou immunitaires in vivo dans des modèles expérimentaux ou in vitro sur des cellules en culture. L’objectif de ces recherches est de comprendre par quel mécanisme la protéine mutée peut entrainer la dégénérescence des motoneurones.
L’objectif de Delphine BOHL dans l’équipe de Séverine BOILLEE est de modéliser la maladie grâce à aux cellules souches pluripotentes humaines (iPSC). Cette technologie de pointe permet de générer des motoneurones ou des cellules immunitaires à partir de cellules de peau (biopsie) de patients. Les iPSC ont deux grandes capacités qui sont celles d’être capables de se multiplier à l’infini et de se différencier en n’importe quel type cellulaire de l’organisme pour peu qu’elles soient exposées aux bons signaux. Ces nouveaux modèles cellulaires sont un outil précieux qui permet pour la première fois d’avoir accès à des neurones humains de patients.
La première étape est de pouvoir caractériser très précisément les motoneurones obtenus à partir des cellules iPSC de patients, d’abord dans des cas génétiques, où la mutation à l’origine de la SLA est connue, puis dans des cas sporadiques afin d’éventuellement identifier des mécanismes communs. La deuxième étape est de réunir dans un même milieu, très contrôlé, les motoneurones et les cellules immunitaires pour modéliser leurs interactions. À termes, ces modèles permettraient également de tester l’efficacité des molécules thérapeutiques.

Les symptômes et la progression de la maladie :
La SLA se caractérise par une paralysie complète des muscles des bras, des jambes et de la gorge entrainant une incapacité à marcher, manger, parler ou même respirer qui s’installe progressivement.
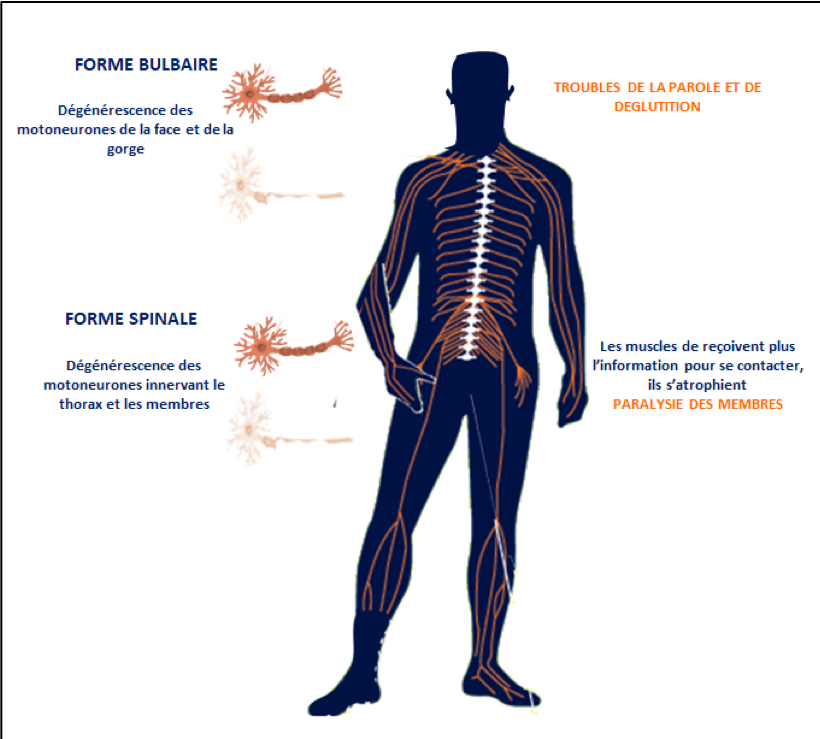
Selon la localisation des motoneurones touchés au début de la maladie, les symptômes initiaux différent :
- Dans environ 30% des cas, la maladie atteint les motoneurones du tronc cérébral (forme bulbaire) et les premières manifestations sont des difficultés à articuler ou à déglutir.
- Dans les autres cas, la maladie touche d’abord les motoneurones périphériques (forme spinale) et débute par une faiblesse et unegêne au niveau d’un membre.La maladie s’intensifie progressivement avec des contractures, une raideur musculaire et des articulations, puis l’atteinte gagne d’autres muscles. Une atrophie musculaireet des troubles de la coordinationgênent la marche et l’habileté manuelle. Les difficultés à déglutir ou à articuler croissent. L’atteinte des muscles respiratoiresintervient souvent à un stade avancé de la maladie et constitue la cause mortelle de la maladie.
A L’Institut du Cerveau – ICM
Séverine BOILLEE étudie l’influence des cellules immunitaires (ou cellules de l’inflammation) sur la dégénérescence des motoneurones et la progression de la maladie. Dans la SLA, les cellules del’inflammation sont beaucoup plus présentes autour du motoneurone quechezlessujetsnonmalades.Ellesréagissentàla dégénérescencedesmotoneuronesetsontimpliquées danslaprogressiondelamaladie.Au niveau du cerveau et de la moelle épinière, lesmotoneuronesinteragissent avec les cellules microgliales alors que dans le système nerveux, périphérie ils sont en contact avecles macrophages. Ces deux typesdecellulesvontjouerundoublerôle:positif,enenvoyant des facteurs bénéfiques pour la survie des motoneurones et négatif via des facteurs toxiques qui vont contribuer à leur destruction.
Les travaux de Séverine BOILLEE visent à mieux comprendre le rôle de ces cellules dans le développement et la progression de la maladie, afin d’identifier de nouvelles pistes thérapeutiques. L’objectif est d’analyser précisément les différents facteurs émis par ces cellules afin d’identifier ceux sur lesquels agir pour ralentir la progression de la maladie.
Une étude promue par l’AP-HP et menée à l’Institut du Cerveau et de la Moelle Épinière à l’Hôpital de la Pitié-Salpêtrière par Isabelle LE BER dans l’équipe d’Alexandra DURR et de Giovanni STEVANIN et par Olivier COLLIOT co-chef d’équipe avec stanley DURRLEMAN a montré, pour la première fois, que des individus asymptomatiques risquant de développer une dégénérescence fronto-temporale (DFT) ou une Sclérose Latérale Amyotrophique (SLA), car porteurs de la mutation c9orf72, présentent des altérations cognitives, anatomiques et structurelles très précoces, avant l’âge de 40 ans. La mise en évidence de biomarqueurs à des stades très précoces est un premier pas vers le développement d’outils nécessaires à l’évaluation de nouveaux traitements. En effet, le développement de thérapeutiques à la phase précoce, idéalement avant le début des symptômes, nécessite de développer des outils qui permettent de savoir quand initier les traitements et de mesurer leur efficacité.
Le diagnostic et les traitements:
Le diagnostic de la SLA est un diagnostic différentiel c’est-à-dire après l’élimination des autres pathologies du système nerveux.
L’examen neurologique associé à un une prise de sang, à un électromyogrammeet une IRMpermettent de confirmer le diagnostic si les symptômes persistent et s’aggravent en quelques semaines.
A ce jour, il n’existe aucun traitement curatif de la SLA.
La prise en charge multidisciplinaire des patients visent à diminuer et soulager les symptômes.
La constitution de centres experts et de réseaux de soin permet cette prise en charge globale selon les besoins des patients.
Le Dr François SALACHAS, cliniciens chercheur dans l’équipe de Séverine BOILLEE à l’Institut du Cerveau – ICM est le coordinateur du centre expert SLA de la Pitié Salpetrière.
A L’Institut du Cerveau – ICM
Concernant les traitements de la SLA, Françoise PIGUET dans l’équipe de Nathalie CARTIER à l’Institut du Cerveau – ICM porte un projet de thérapie génique et cellulaire pour la SLA. L’équipe étudie des modèles expérimentaux afin d’optimiser la distribution de gènes médicaments et d’évaluer la sécurité et l’efficacité de ces approches avant d’initier des essais thérapeutiques.