





Plusieurs avancées majeures sur les “ataxies spinocérébelleuses” (ou SCA pour SpinoCerebellar Ataxia), marquent l’actualité scientifique de l’Institut du Cerveau – ICM en faisant l’objet de 2 publications dans Brain et d’une 3ème dans The American Journal of Human Genetics.
Maladie neurodégénérative du cervelet se manifestant par des symptômes moteurs spécifiques tels que les troubles de l’équilibre, les SCAs font partie des maladies rares et sont de fait difficiles et complexes à étudier et à comprendre. Les avancées réalisées et mises en lumière dans ces 3 articles, sont le résultat de projets internationaux de grande envergure dans lesquels se sont impliqués les chercheurs de l’Institut du Cerveau – ICM. Ces nouvelles avancées font suite à la découverte d’un gène responsable de la maladie.
QU’EST-CE QUE L’ATAXIE SPINOCÉRÉBELLEUSE (SCA) :
Les ataxies (du grec ataxiā, signifiant « désordre ») aussi appelées syndromes cérébelleux, sont un ensemble de pathologies neurodégénératives du cervelet et/ou du tronc cérébral affectant spécialement l’équilibre, la marche et les mouvements oculaires.
Touchant le cervelet (le “petit cerveau” situé à la base arrière du cerveau), partie du système nerveux central qui assure le maintien de la posture et de l’équilibre et qui coordonne les mouvements volontaires, les ataxies sont des pathologies neuromusculaires qui ne sont pas liées à une déficience physique des muscles mais à une neurodégénérescence du système nerveux cérébelleux ou de ses connections. Ainsi, lorsque le cervelet ne peut plus assurer ses fonctions, les symptômes propres aux ataxies apparaissent et évoluent de manière plus ou moins rapide.
Les ataxies font parties des maladies rares neurogénétiques (maladie du système nerveux qui touche l’ADN) qui peuvent se déclencher chez des sujets de tous âges. Plusieurs types d’ataxies peuvent être distingués selon leur mode de transmission génétique (dominant ou récessif).
Les ataxies spinocérébelleuses sont des ataxies cérébelleuses de transmission dominante (il suffit d’être porteur de l’anomalie génétique pour développer la maladie), à la différence des ataxies cérébelleuses de transmission récessive dont la forme la plus fréquente est l’ataxie de Friedreich.
Les ataxies spinocérébelleuses ou SCA (pour SpinoCerebellar Ataxia) touchent 1 à 5 personnes sur 100 000 et apparaissent en moyenne vers l’âge de 35 ans. Elles se manifestent cliniquement et génétiquement de façon très hétérogène. Les chercheurs ont jusque-là identifié une 30aine de gènes différents impliqués dans le développement de la maladie. Les chercheurs s’attellent à comprendre le lien entre ces différentes mutations et l’âge d’apparition des symptômes ainsi que la sévérité des SCAs.
IDENTIFICATION DE DEUX NOUVEAUX GÈNES IMPLIQUÉS DANS LES SCAS
(Delplanque et al., Brain, 2014) et (Di Gregorio et al., Am J Hum Genet, 2014)
Alexandra Durr (Professeur Sorbonne Université, praticien APHP) et Giovanni Stevanin de l’Institut du Cerveau – ICM (DR Inserm et Professeur EPHE) et leurs collègues du réseau international SPATAX ont contribué à l’identification de 2 nouveaux gènes dont les mutations sont responsables de formes autosomiques dominantes d’ataxie spinocérébelleuse. Ces maladies neurologiques qui affectent le cervelet, parfois très invalidantes et létales, sont expliquées dans 50% des cas par des mutations dans une trentaine de gènes mais il reste encore beaucoup de cas non expliqués.
Les équipes du réseau SPATAX, notamment à Lille (Pr Bernard Sablonnière), Turin (Dr Alfredo Brusco) et à l’Institut du Cerveau – ICM (Pr Giovanni Stevanin) ont recherché les gènes en cause dans 2 grandes familles par une approche combinant la cartographie génétique et le séquençage nouvelle génération de l’exome. Les résultats de ces approches viennent d’être publiés en juillet et aout.

L’un de ces gènes (ELOVL5/SCA38) code pour un enzyme du métabolisme lipidique ouvrant la possibilité d’un diagnostic par dosage des acides gras polyinsaturés dans le plasma des patients. Les mutations de ce gène entrainent l’accumulation de cet enzyme dans le compartiment cellulaire du Golgi.
Le second gène (TMEM240/SCA21) code pour une protéine membranaire abondante au niveau des synapses neuronales mais de rôle inconnu. Les mutations de ce gène sont associées à des phénotypes sévères incluant une atteinte cognitive ou un retard mental d’évolution lente.
La découverte de ces gènes représente un nouvel espoir pour déterminer de nouvelles cibles thérapeutiques potentielles.
MISE EN ÉVIDENCE D’UNE CORRÉLATION
Il semblerait exister une corrélation entre l’âge d’apparition des SCAs et la répétition d’une séquence ADN de 3 nucléotides (CAG) dans différents gènes (Tezenas du Montcel et al., Brain, 2014)
Une vaste étude internationale pilotée par l’équipe d’Alexis Brice à l’Institut du Cerveau – ICM et notamment par Giovanni Stevanin (chercheur Inserm et Professeur EPHE) et Sophie Tezenas du Montcel (APHP) vient d’être publiée dans le journal BRAIN et l’article a été choisi par l’Éditeur comme article du mois de septembre. Il s’agit de l’étude comparative des données génétiques d’une cohorte de taille record de plus de 1900 patients atteints de SCA, dans l’objectif d’en dégager les caractéristiques moléculaires de la maladie. Ce projet d’envergure s’est concentré sur des patients dont les SCAs sont dues à des mutations par “expansion de trinucléotides” dans les gènes : répétitions d’une séquence ADN de trois nucléotides « CAG » ((CAG)n, codant pour une polyglutamine.

Cette étude internationale (projets EUROSCA et NEUROMICS) associant 12 pays démontre que la taille de l’expansion dans les gènes mutés dans les SCAs n’explique qu’une partie de l’âge de début des symptômes et que les répétitions trinucléotidiques normales dans d’autres gènes participent également à la variance de l’âge d’apparition. Cette découverte est donc un pas supplémentaire dans la compréhension des SCAs et l’identification de tous les gènes qui peuvent moduler la sévérité de la pathologie (âge de début, signes associés, vitesse d’aggravation des signes…) pourraient permettre de mettre en place des stratégies thérapeutiques alternatives visant à ralentir la progression de la maladie.
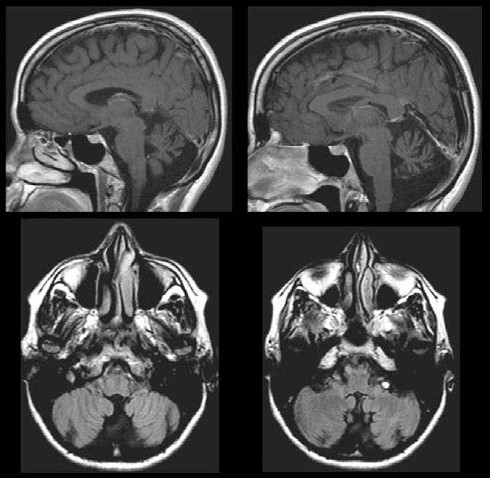
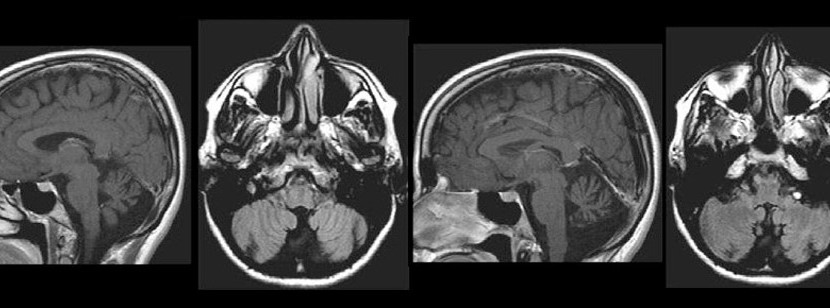
IRM cérébrale d’un patient atteint d’ataxie spinocérébelleuse, muté pour le gène ELOVL5. On peut noter l’atrophie du vermis du cervelet alors que le cortex cérébral et le tronc cérébral sont préservés (Figure 2.A extraite de Di Gregorio et al., Am J Hum Genet, 2014).
