





Et si un ver permettait un bond en avant dans la recherche sur les maladies à prions comme la maladie de Creutzfeldt-Jakob ? Un travail récent du groupe de Nicolas Bizat (Université de Paris) et Stéphane Haïk (Inserm) à l’Institut du Cerveau, publié dans la prestigieuse revue Brain, suggère le potentiel de ce modèle pour la recherche thérapeutique et identifie cinq molécules prometteuses dans ces pathologies.
Les maladies à prions, dont fait partie la maladie de Creutzfeldt-Jakob, sont des affections neurodégénératives fatales. Elles se caractérisent par une agrégation pathologique de la protéine prion. Lorsque cette protéine est mal conformée, elle forme des assemblages dans les neurones conduisant à leur mort. Cette mauvaise conformation de la protéine a par ailleurs la capacité de se propager de neurones en neurones. L’origine des maladies à prions est diverse, pouvant être infectieuses, génétique ou sporadique (sans source identifiée). Il n’existe à l’heure actuelle aucun traitement pour ces maladies, ni pour retarder leur apparition ou ralentir leur progression.
Pour en savoir plus sur les maladies à prions : https://institutducerveau-icm.org/fr/maladie-prion-creutzfeldt-jakob/
Les difficultés de la recherche de thérapies dans les maladies à prions
Un enjeu clé de la recherche sur les maladies à prions est de développer de nouveaux modèles in vivoexpérimentaux permettant des analyses à haut-débit de composés ayant potentiellement la capacité de lutter contre l’agrégation pathologique de la protéine prion ou d’agir sur la survie des neurones. Il n’existe à l’heure actuelle que très peu de modèles cellulaires ou in vivo récapitulant les caractéristiques de la maladie ayant pour origine une mutation génétique.
Parmi les formes génétiques de la maladie de Creutzfeldt-Jakob, la plus commune est due à la mutation E200K du gène codant pour la protéine prion. L’identification de cette mutation permet aujourd’hui de développer de nouveaux modèles génétiques d’étude de ces pathologies dont les mécanismes cellulaires toxiques demeurent encore mal compris à ce jour.
Le nématode Caenorhabditis elegans, un modèle in vivo prometteur
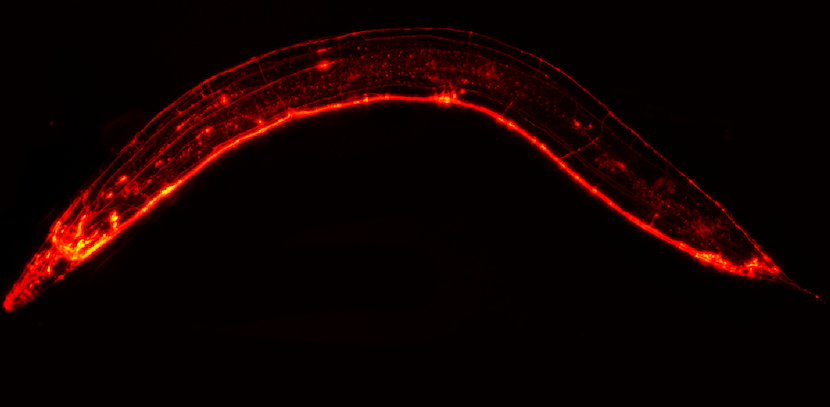
Nicolas Bizat (Université de Paris), du groupe de Stéphane Haïk (Inserm) à l’Institut du Cerveau, a mis au point un nouveau modèle génétique d’étude du prion, grâce au nématode Caenorhabditis elegans, un ver rond transparent de petite taille. Il présente plusieurs avantages importants comme le fait d’être transparent, de posséder un système neuronal caractérisé, et surtout, de ne pas être pourvu d’un homologue de la protéine prion présente chez l’humain.
Les chercheurs ont ainsi pu exprimer la protéine prion avec la mutation E200K dans le système nerveux de cet organisme et en étudier ses répercussions. Cela a eu pour conséquence chez ces nématodes de l’apparition de comportements neuro-fonctionnels spécifiques, l’accumulation d’inclusions d’agrégats de protéine prions dans les neurones ainsi qu’à une importante neurodégénérescence.
Fort de ce nouveau modèle reproduisant des caractéristiques clés de la pathologie à l’échelle moléculaire, les chercheurs de l’Institut du Cerveau ont testé le pouvoir « anti-prion » de plus de 320 composés déjà validés pour une utilisation thérapeutique chez l’humain et connus pour leur capacité à atteindre facilement le cerveau. Ils ont identifié 5 molécules efficaces sur l’agrégation de la protéine, la fonctionnalité ainsi que la dégénérescence des neurones atteints dans leur nouveau modèle génétique d’expression de la protéine prion pathologique chez le nématode Caenorhabditis elegans.
Grâce à cet outil expérimental, les chercheurs de l’Institut du Cerveau apportent aujourd’hui une solution aux limitations existant pour le criblage à haut-débit de molécules au potentiel thérapeutique, tout en proposant un nouveau modèle in vivo pour l’étude des mécanismes pathologiques associés aux maladies à prions et, peut-être plus généralement, à d’autres maladies neurodégénératives impliquant des protéines pouvant s’agréger et se propager dans le système nerveux central comme les maladies d’Alzheimer et de Parkinson. Les 5 molécules identifiées vont à présent pouvoir être testées sur des modèles plus complexes d’infections au prion et d’autres modèles de protéinopathies afin de confirmer leurs effets thérapeutiques.
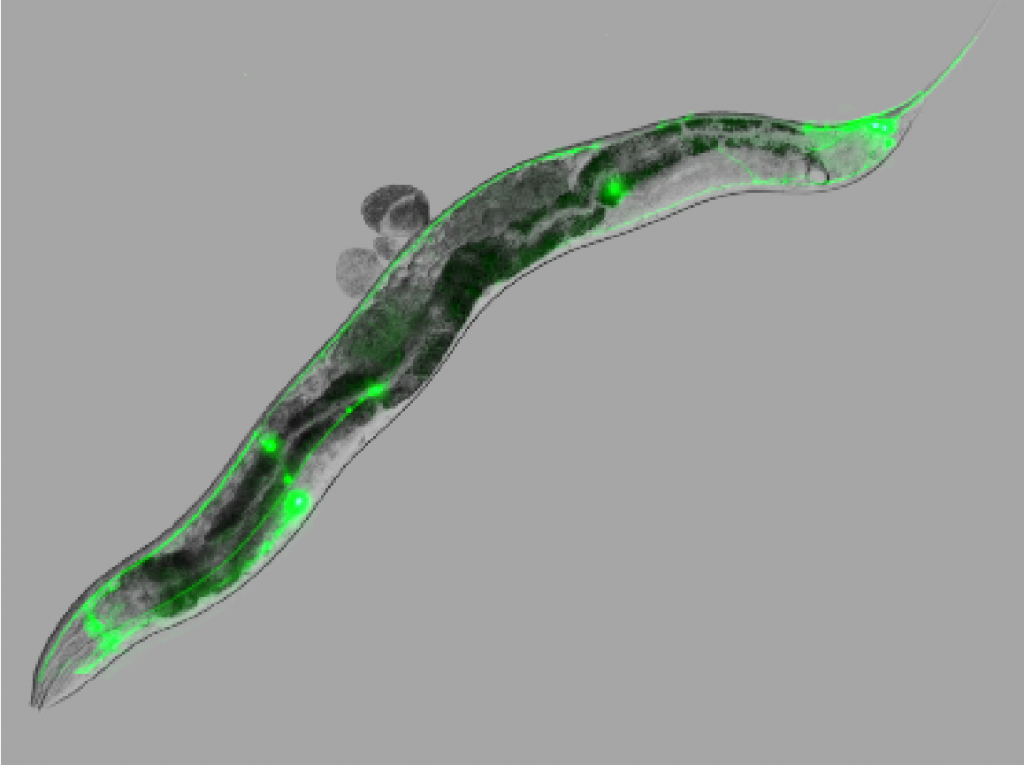
Image d’un nématode (C. elegans) exprimant la protéine prion humaine dans le système mécanosensitif (vert)
Source
An in vivo Caenorhabditis elegans model for therapeutic research in human prion diseases. Bizat N, Parrales V, Laoues S, Normant S, Levavasseur E, Roussel J, Privat N, Gougerot A, Ravassard P, Beaudry P, Brandel JP, Laplanche JL, Haïk S.Brain. 2021 Oct 22;144(9):2745-2758.