





Les dysfonctionnements des canaux ioniques – ou canalopathies – dans le cerveau sont aujourd’hui associés à plus de 30 maladies neurologiques comme l’épilepsie ou encore les ataxies cérébelleuses. Structures situées sur la membrane des cellules permettant le passage d’ions (par exemple les ions sodium et potassium) entre l’intérieur d’une cellule et son environnement extérieur (milieu extracellulaire), ces canaux permettent notamment de générer et contrôler les potentiels d’action dans les neurones. Une étude menée à l’Institut du Cerveau (Sorbonne Université/Inserm/AP-HP/CNRS) a permis d’identifier une nouvelle canalopathie cérébrale ayant pour origine des mutations dominantes du gène KCNN2, codant pour le canal ionique SK2. Les résultats ont été publiés dans Brain le 27 novembre 2020.
Le potentiel d’action est un message de nature électrique qui transmet l’information d’un neurone à l’autre ou à une cellule effectrice comme par exemple les cellules musculaires via la synapse.
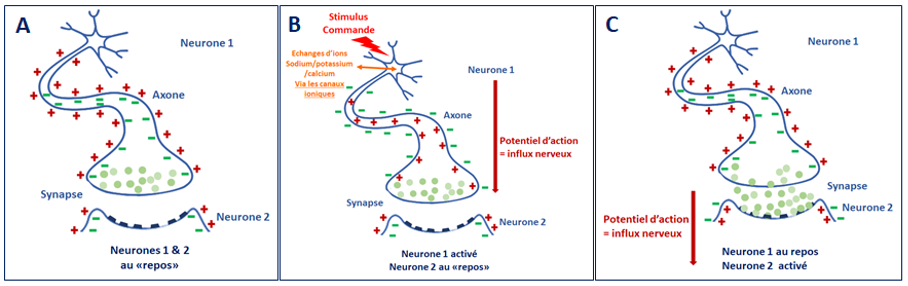
Lors du passage du potentiel d’action, on observe une inversion des charges électriques entre l’extérieur et l’intérieur de la cellule appelée dépolarisation. Après le passage de l’influx nerveux le neurone revient à un état de repos appelé la repolarisation. Les canaux ioniques permettent notamment les échanges des ions sodium et potassium qui entraînent ces changements de polarisation de la cellule.
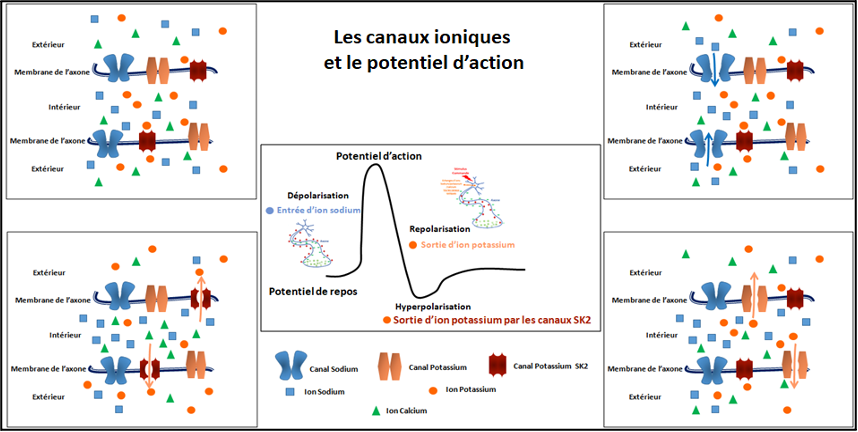
Les canaux ioniques SK2, codés par le gène KCNN2, sont exprimés uniquement dans les neurones et permettant le passage des ions potassium selon le taux d’ions calcium à l’intérieur du neurone. Ils jouent un rôle important dans la régulation du potentiel d’action en contrôlant la durée de la phase de retour à un potentiel de repos des neurones. Le blocage pharmacologique des canaux SK2 dans des modèles expérimentaux entraine des troubles de l’apprentissage et de la mémoire.
Le Dr Fanny Mochel, généticienne au sein du département de génétique de l’hôpital de la Pitié-Salpêtrière AP-HP et chercheuse à l’Institut du Cerveau (Sorbonne Université/Inserm/AP-HP/CNRS) et le Pr Christel Depienne, généticienne à l’institut de génétique humaine de l’Hôpital Universitaire d’Essen (Allemagne) et également chercheuse à l’Institut du Cerveau ont identifié un nouveau syndrome associé à des mutations du canal SK2. L’étude publiée dans la revue scientifique Brain porte sur 10 patients, 6 hommes et 4 femmes âgés de 2 à 60 ans présentant des retards intellectuels plus ou moins sévères associés, pour certains, à des troubles du spectre autistique ou des épisodes psychotiques. Ces troubles cognitifs sont dans tous les cas associés à des tremblements, ou des symptômes d’ataxie cérébelleuse ou encore des mouvements anormaux.
Grâce à une collaboration avec Agnes Rastetter de la plateforme de génotypage/séquençage (iGenSeq) de l’Institut du Cerveau (Sorbonne Université/Inserm/AP-HP/CNRS), le génome d’un premier patient recruté à la Pitié-Salpêtrière a été analysé à la recherche de mutations génétiques à l’origine de ce syndrome. Cette analyse a mis en évidence une mutation du gène KCNN2 interrompant sa séquence codante, absente des parents du patient (mutation de novo). L’imagerie cérébrale par IRM (imagerie par résonance magnétique) chez ce patient a mis en évidence des anomalies de structure et d’intégrité de la substance blanche du cerveau, c’est-à-dire la gaine cérébrale protectrice des axones des neurones.
Par ailleurs, une collaboration internationale a permis aux chercheurs d’identifier 9 autres patients avec mutations du gène KCNN2. La majorité de ces mutations étaient survenues de novo tandis qu’une mutation étaient transmises dans une forme familiale du même syndrome.
Enfin, en travaillant conjointement avec Carine Dalle de la plateforme d’exploration cellulaire d’électrophysiologie (CELIS ePHYS) de l’Institut du Cerveau, les équipes des Dr Mochel et Depienne, ont montré un rôle délétère de ces mutations sur la fonction du canal SK2, c’est-à-dire une perte de fonction entrainant un dysfonctionnement du canal ionique SK2 et donc une perte de régulation du potentiel d’action, support du message nerveux entre neurones et entre les neurones et les muscles.
Les résultats de cette nouvelle étude ont permis d’identifier une nouvelle canalopathie cérébrale ayant pour origine des mutations dominantes du gène KCNN2, codant pour le canal ionique SK2. Ce nouveau syndrome se caractérise par la présence d’une part, de symptômes cognitifs, et en particulier une déficience intellectuelle, et d’autre part, de symptômes moteurs tels que des mouvements anormaux.
Cette nouvelle pathologie, dont on connaît maintenant la cause, est très hétérogène d’un point de vue des symptômes et nécessite une prise en charge multidisciplinaire à la frontière entre la génétique, pour la recherche des mutations du gène KCNN2, la neuropédiatrie et neurologie pour la prise en charge des manifestations cognitives et motrices des patients.
Référence:
Variants in the SK2 channel gene (KCNN2) lead to dominant neurodevelopmental movement disorders, Fanny Mochel, Agnes Rastetter, Berten Ceulemans, Konrad Platzer, Sandra Yang, Deepali N. Shinde, Katherine L. Helbig, Diego Lopergolo, Francesca Mari, Alessandra Renieri, Elisa Benetti, Roberto Canitano, Quinten Waisfisz, Astrid S. Plomp, Sylvia A. Huisman, Golder N. Wilson, Sara S. Cathey, Raymond J. Louie, Daniela Del Gaudio, Darrel Waggoner, Shawn Kacker, Kimberly M. Nugent, Elizabeth R. Roeder, Ange-Line Bruel, Julien Thevenon, Nadja Ehmke, Denise Horn, Manuel Holtgrewe, Frank J. Kaiser, Susanne B. Kamphausen, Rami Abou Jamra, Sarah Weckhuysen, Carine Dalle and Christel Depienne.